Swiss Institute of Bioinformatics
Click2Drug
Directory of computer-aided Drug Design tools
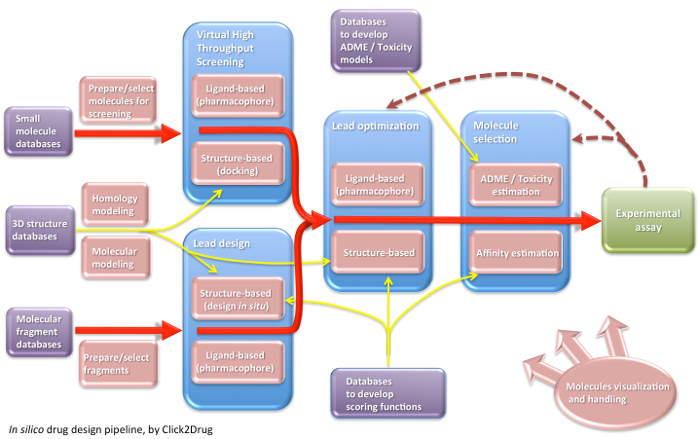
Click2Drug contains a comprehensive list of computer-aided drug design (CADD) software, databases and web services. These tools are classified according to their application field, trying to cover the whole drug design pipeline. If you think that an interesting tool is missing in this list, please contact usClick on the following picture to select tools related to a given activity:
Show all links Hide all links
Databases
Databases handling
- JChem for Excel. Integrates structure handling and visualizing capabilities within a Microsoft Excel environment. Structures are fully supported within spreadsheets and be can viewed, edited, searched, resized, ordered, managed. Provided by ChemAxon.
- ChemDiff. Indigo-based utility for finding duplications and visual comparison of two files containing multiple structures. SDF, SMILES, CML, MOLFILE input formats are supported. Files can contains large amount of molecules and ChemDiff was test on files with up to 1 million ones. Free and open-source. Distributed by GGA software.
Chemical structure representations
2D drawing
- ChemDraw. Molecule editor developed by the cheminformatics company CambridgeSoft. For Windows and Mac.
- MarvinSketch. Advanced chemical editor for drawing chemical structures, queries and reactions developed by ChemAxon. Exists as an applet.
- ACD/ChemSketch. Molecule editor developed by ACD/Labs. Also available as freeware, with tools for 2D structure cleaning, 3D optimization and viewing, InChI generation and conversion, drawing of polymers, organometallics, and Markush structures. For Windows only.
- DataWarrior. Free Cheminformatics Program for Data Visualization and Analysis. DataWarrior combines dynamic graphical views and interactive row filtering with chemical intelligence. Scatter plots, box plots, bar charts and pie charts not only visualize numerical or category data, but also show trends of multiple scaffolds or compound substitution patterns. Compounds can be clustered and diverse subsets can be picked. Calculated compound similarities can be used for multidimensional scaling methods, e.g. Kohonen nets. Physicochemical properties can be calculated, structure activity relationship tables can be created and activity cliffs be visualized.
- JKluster. Tool of JChem for clustering, diversity calculations, and library comparisons based on molecular fingerprints and other descriptors. Useful in combinatorial chemistry, drug design, or other areas where a large number of compounds need to be analyzed. Provided by ChemAxon.
- SMARTSeditor. Graphic editing tool for generic chemical patterns. Based on the SMARTS language, chemical patterns can be created and edited interactively, similar to molecule editing in a chemical structure editor. The visualization of patterns is based on the visualization concept of the SMARTSviewer.Freely available for linux systems with 32 and 64 bit, windows 32bit systems and MacOS. Developed by the University of Hamburg.
- VLifeBase. Provides features to build a molecule from scratch using 2D Draw and conversion to 3D. The 3D editor allows addition, modification, replacement and deletion of atoms, bonds and groups, with Undo and Redo operations. Provided by VLife.
- ISIS/Draw. Chemical structure drawing program for Windows, published by MDL Information Systems. Free of charge for academic and personal use.
- ChemDoodle. Chemical structure environment with a main focus on 2D graphics and publishing to create media for structures, reactions and spectra. For Windows, Mac and Linux.
- ChemDraw for iPad. iPad application to create, edit and share publication-quality chemical structures with just the touch of a finger, based on the world’s most popular chemical drawing software, ChemDraw. Provided by PerkinElmer, Inc.
- TouchMol Deskop Application. Tool for drawing chemical and biological structures, optimized for Touch Operations. Allows Copy/Paste to ChemDraw, ISIS/Draw, SciFinder and Word. Provides name-to-structure. For Windows 8. Provided by Scilligence.
- TouchMol for Office. Desktop tool for drawing chemical and biological structures, into the MS Office suite. Provided by Scilligence.
- ChemDoodle Mobile. Free iPhone companion to ChemDoodle. ChemDoodle Mobile is a calculator for drawn organic structures. There are four main windows: Draw, Calculate, Spectra and Help. The Draw window shows a typical ChemDoodle sketcher, where you can draw and store your structures. The Calculate page calculates properties and the Spectra page simulates NMR spectra. All spectra are interactive. The Help page contains a thorough help guide. Provided by iChemLabs.
- Chirys Draw. Application for drawing publication-quality molecular structures and reactions. Designed from the ground up for the iPad. Developed by Integrated Chemistry Design, Inc.
- Chirys Sketch. Application for drawing publication-quality molecular structures and reactions, for iPhone and iPod Touch. Developed by Integrated Chemistry Design, Inc.
- Mobile Molecular DataSheet. Allows viewing and editing chemical structure diagrams on an iPhone, iPod or iPad. Molecules are organized in collections of datasheets. Individual molecules, or whole datasheets, can be shared via iTunes or sent by email, using the standard MDL MOL and SDfile formats, which allows the data to be integrated into any external workflow. Provided by Molecular Materials Informatics, Inc.
- SAR Table. Application designed for creating tables containing a series of related structures, their activity/property data, and associated text. Structures are represented by scaffolds and substituents, which are combined together to automatically generate a construct molecule. The table editor has many convenience features and data checking cues to make the data entry process as efficient as possible. For iPad. Provided by Molecular Materials Informatics, Inc.
- Molprime+. Chemical structure drawing tool based on the unique sketcher from the Mobile Molecular DataSheet. Can send structure data via email, open structures from email or web, create graphical images or Microsoft Word documents with embedded structure graphics, calculate properties based on structures and use structures to search Mobile Reagents and ChemSpider. Provided by Molecular Materials Informatics, Inc.
- StructureMate. Portable chemical dataset viewer for iPad, for browsing SAR reports, chemical catalogs, custom-made databases, and physical property references. Provided by Metamolecular, LLC.
- Elemental. Chemistry sketch for iphone and ipad. Developed by Dotmatics Limited.
- Accelrys Draw. Allows drawing and editing complex molecules, chemical reactions and biological sequences. provided by Accelrys.
- PLT. Program for producing chemical drawings and outputting them in a variety of formats. For Windows.
- JChemPaint. Free and open source editor and viewer for chemical structures in 2D. Exists as a Java stand alone application and two varieties of Java applet that can be integrated into web pages. Platform-independent.
- BKchem. BKChem is a free open source chemical drawing program written in Python. Platform-independent.
- MolSketch. Free open source molecular drawing tool for 2D molecular structures. Available for Windows, Mac and Linux.
- JME Molecular Editor. Java applet which allows to draw / edit molecules and reactions (including generation of substructure queries) and to depict molecules directly within an HTML page. Editor can generate Daylight SMILES or MDL Molfile of created structures.
- Chem4D. Molecular drawing tool. Includes assignment of systematic names to organic structures according to IUPAC nomenclature rules, and drawing of molecules from IUPAC names. For Windows and Mac. Distributed by ChemInnovation Software.
- XDrawChem. Free open source software program for drawing chemical structural formulas, available for Windows, Unix, and Mac OS.
- iMolecular Draw. Application that can view, edit and build molecules in 2D. For iPhone.
- SketchEl. Free and open source interactive chemical molecule sketching tool, and molecular spreadsheet data entry application. Written in Java. Exists as an applet.
- Chemtool. Free open source program for drawing chemical structures on Linux and Unix systems using the GTK toolkit under X11.
- Bioclipse. Java-based, open source, visual platform for chemo- and bioinformatics based on the Eclipse Rich Client Platform (RCP).
- Chrawler. Can scan all data sources, including local files, remote files on network, emails, web pages, SharePoint contents, etc., and find contained chemical structures, and make them structure-searchable (substructure, full-structure, similarity). Distributed by Scilligence.
- Imago. Toolkit for 2D chemical structure image recognition. It contains a GUI program and a command-line utility, as well as a documented API for developers. Imago is completely free and open-source, while also available on a commercial basis. Distributed by GGA software.
- Imago OCR Visual Tool. Java GUI for Imago. Ego is completely free and open-source, while also available on a commercial basis. Distributed by GGA software.
- Imago Console Application. Command-line interface for Imago. Alter-Ego is completely free and open-source, while also available on a commercial basis. Distributed by GGA software.
- OLN Chem4SharePoint. Makes it possible to draw, display and search chemical structures in SharePoint. Distributed by Scilligence.
- ChemJuice. Molecular drawing software for iPhone. Developed by IDBS.
- ChemJuice Grande. Molecular drawing software for iPad. Developed by IDBS.
- MolPad. Free chemical structure drawing application. It can draw structures from scratch or load them from ChemSpider and modify them. Structures can be emailed in Molfile format. For Android.
- DCE ChemPad. Free application to draw chemical structures and calculate molecular weight, molecular formula and to send the molfile. It shows the capabilities of the Dendro Chemical Editor control for Android to build chemistry-aware mobile applications. For Android.
- Indigo-depict. Command-line molecule and reaction rendering utility. Free and open source. Distibuted by GGA software.
2D drawing online
- jsMolEditor. Molecule Editor in JavaScript. Open source.
- Marvin molecule editor and viewer. Java based chemical editor for drawing chemical structures. Includes unlimited structure based predictions for a range of properties (pKa, logD, name<>structure, etc.). Provided by ChemAxon.
- Ketcher. Web-based chemical structure editor written in JavaScript. Free and open-source, but also available on a commercial basis. Distributed by GGA software.
- ChemWriter. Chemical structure editor designed for use with Web applications. Distributed by Metamolecular.
- Molinspiration WebME Molecule Editor. Allows creation and editing of molecules in browsers without Java support and without any plugins. The editor is based on a Web2.0 Ajax technology. WebME allows therefore web-based structure input also in institutions where Java applets are not allowed and offers complete platform compatibility. The actual molecule processing in WebME is based on the JMEPro editing engine running on a server. provided by Molinspiration.
- OLN JSDraw. Javascript libary you can display and draw chemical structures in web pages, which works cross browser, including IE, Firefox, Safari, Opera and Chrome, crose platform, including Window, Mac, Linux, and even iPhone, Android and other mobile devices. Free for education. Provided by Scilligence.
- TouchMol Web. Tool for drawing chemical and biological structure online. Allows Copy/Paste to ChemDraw, ISIS/Draw, SciFinder and Word. Provides name-to-structure. Provided by Scilligence.
3D viewers
- UCSF Chimera. Open source, highly extensible program for interactive visualization and analysis of molecular structures and related data. Free of charge for academic, government, non-profit, and personal use. For Windows, Mac and Linux. Developed by the Resource for Biocomputing, Visualization, and Informatics, UCSF.
- Pymol. Open source, user-sponsored, molecular visualization system written in Python. Distributed by DeLano Scientific LLC. For Windows, Mac and Linux.
- OpenStructure. Open-source, modular, flexible, molecular modelling and visualization environment. It is targeted at interested method developers in the field of structural bioinformatics. Provided by the Swiss Institute of Bioinformatics and the Biozentrum, University of Basel.
- Swiss-PDB Viewer / DeepView. Program for 3D visualization of macromolecules, allowing to analyze several proteins at the same time. Swiss-PdbViewer is tightly linked to SWISS-MODEL, an automated homology modeling server developed within the Swiss Institute of Bioinformatics (SIB).
- Computer-Aided Drug-Design Platform using PyMOL. PyMOL plugins providing a graphical user interface incorporating individual academic packages designed for protein preparation (AMBER package and Reduce), molecular mechanics applications (AMBER package), and docking and scoring (AutoDock Vina and SLIDE).
- Computer-Aided Drug-Design Platform using PyMOL. a simple Java tool for visual exploration of three-dimensional (3D) virtual screening data. The VSviewer3D brings together the ability to explore numerical data, such as calculated properties and virtual screening scores, structure depiction, interactive topological and 3D similarity searching, and 3D visualization. By doing so the user is better able to quickly identify outliers, assess tractability of large numbers of compounds, visualize hits of interest, annotate hits, and mix and match interesting scaffolds. We demonstrate the utility of the VSviewer3D by describing a use case in a docking based virtual screen. Developed by Data2Discovery Consulting Inc., USA.
- Autodock Vina plugin for PyMOL. Allows defining binding sites and export to Autodock and VINA input files, doing receptor and ligand preparation automatically, starting docking runs with Autodock or VINA from within the plugin, viewing grid maps generated by autogrid in PyMOL, handling multiple ligands and set up virtual screenings, and set up docking runs with flexible sidechains.
- Dehydron. A dehydron calculator plugin for PyMOL. This plugin calculates dehydrons and display them onto the protein structure.
- pymacs. Python module for dealing with structure files and trajectory data from the GROMACS molecular dynamics package. It has interfaces to some gromacs functions and uses gromacs routines for command line parsing, reading and writing of structure files (pdb,gro,...) and for reading trajectory data (only xtc at the moment).
- PyRosetta. Interactive Python-based interface to the Rosetta molecular modeling suite. It enables users to design their own custom molecular modeling algorithms using Rosetta sampling methods and energy functions.
- Visual Molecular Dynamics (VMD). Free open source molecular visualization program for displaying, animating, and analyzing large biomolecular systems using 3-D graphics and built-in scripting. For MacOS X, Unix, or Windows. Developed by the NIH resource for macromolecular modeling and bioinformatics, University of illinois.
- ePMV. (embedded Python Molecular Viewer). Free, open-source plug-in that runs molecular modeling software directly inside of professional 3D animation applications (hosts, i.e. Blender, Cinema4D and Maya 2011) to provide simultaneous access the capabilities of all of the systems. Developed by the Scripps Research Institute.
- Jmol. Open source Java viewer for chemical structures in 3D.
- Zodiac. Free open source molecular modelling suite for computation, analysis and display of molecular data. It features state-of-the-art tools for managing molecular databases, run molecular docking experiments, compute raytraced images, etc... Developed by Zeden. For windows, Mac and Linux.
- GLmol. Free and open source 3D molecular viewer based on WebGL and Javascript. GLmol runs on newer versions of Firefox, Chrome, Safari or Opera. Internet Explorer is not supported. GLmol also runs on Sony Ericsson's Android devices which support WebGL and WebGL enabled safari in iOS.
- DS Visualizer. Free 3D visualizer of Discovery Studio. Allows sequence handling and, 2D or 3D charting. Creates 2D ligand-receptor interaction diagrams. Distributed by Accelrys. DS Visualizer ActiveX Control allows visualizing and interacting with molecules in Microsoft Office documents and Internet Explorer. For Windows and Linux.
- OpenAstexViewer. Free open source java molecular graphics program that assists in structure based drug design. It can be used as an Applet in a web page or as a desktop application. Provided by Astex Therapeutics. For Windows, linux and Mac.
- ICM-Browser. Free molecular visualization program for displaying proteins, DNA and RNA, and multiple sequence alignments. Allows saving interactive 3D files to display on the web or in PowerPoint. Distributed by Molsoft. For Windows, Mac and linux. Exist in a Pro version.
- Crystal Studio. Crystal Studio is a Windows XP/Vista/Windows 7 (32/64) software package for crystallography. It is a comprehensive tool for user-friendly creation, 3D graphical design, display and manipulation of crystal and macro-molecular structures, surface or interfaces and defects and for the simulation of X-Ray, neutron and electron diffraction patterns.
- Friend. Integrated Front-End application for multiple structure visualization and multiple sequence alignment. Friend is a bioinformatics application designed for simultaneous analysis and visualization of multiple structures and sequences of proteins and/or DNA/RNA. The application provides basic functionalities such as: structure visualization with different rendering and coloring, sequence alignment, and simple phylogeny analysis, along with a number of extended features to perform more complex analyses of sequence structure relationships, including: structure alignment of proteins, investigation of specific interaction motifs, studies of protein-protein and protein-DNA interactions, and protein super-families. Friend is also available as an applet. Provided by the Ray and Stephanie Lane Center for Computational Biology.
- Chemkit. Free open-source C++ library for molecular modelling, cheminformatics, and molecular visualization.
- Coot. Program for macromolecular model building, model completion and validation, particularly suitable for protein modelling using X-ray data. Free and open-source.
- Jamberoo. Free open source program for displaying, analyzing, editing, converting, and animating molecular systems (former JMolEditor). For Windows, Mac and Linux.
- YASARA View. Free molecular visualization program for displaying macromolecules, building molecules, multiple sequence alignments. Can be complemented by YASARA Model. Provided by YASARA.
- QuteMol. Open source (GPL), interactive, high quality molecular visualization system. QuteMol exploits the current GPU capabilites through OpenGL shaders to offers an array of innovative visual effects. QuteMol visualization techniques are aimed at improving clarity and an easier understanding of the 3D shape and structure of large molecules or complex proteins. Developed by the Visual Computing Lab at ISTI-CNR, Italy.
- Molekel. Free open-source multi-platform molecular visualization program, for Mac OSX, Windows and Linux. Provided by the Swiss National Supercomputing Centre: Lugano (Switzerland).
- NOC. Free molecular explorer for protein structure visualization, validation and analysis. Mainained by Dr. Nymeyer's Group, Inst. Mol. Biol., Florida State University.
- CueMol. Program for the macromolecular structure visualization (CueMol was formerly called "Que"). CueMol aims to visualize the crystallographic models of macromolecules with the user-friendly interfaces. Currently supported files are molecular coordinates (PDB format), electron density (CCP4, CNS , and BRIX formats), MSMS surface data, and APBS electrostatic potential map.
- TexMol. Molecular visualization and computation package. Free and open source software.
- Chil2 Viewer. Visualization tool and graphical user interface of the Chil2 suite, with analysis tools, database integration and ruby interface. Open for general research.
- VEGA ZZ. Visualization application and molecular modeling toolkit (Molecular mechanics and dynamics, structure-based screening). Free for non-profit academic uses. Provided by the Drug Design Laboratory of the University of Milano.
- BALLView. Standalone molecular modeling and visualization application. Provides a framework for developing molecular visualization functionality. Can be used as the visualizaion component of BALL. Free and opensource. For Windows, Mac and Linux.
- RasMol. Program for molecular graphics visualisation.
- RasTop. Free open source molecular visualization software adapted from the program RasMol. RasTop wraps a user-friendly graphical interface around the "RasMol molecular engine". Developed for educational purposes and for the analysis of macromolecules at the bench. For Windows and Linux.
- Cn3D. Visualization tool for biomolecular structures, sequences, and sequence alignments. Maintained and distributed by the NCBI. For Windows, Mac and Linux.
- Bodil. Free, modular, multi-platform software package for biomolecular visualization and modeling. Bodil aims to provide easy three-dimensional molecular graphics closely integrated with sequence viewing and sequence alignment editing.
- COSMOS Viewer. Free software for presentation of molecules.
- BARISTA. BARISTA visualization functions create, display, and manipulate 3D depictions of molecular structures based on results computed by molecular computation programs such as Conflex, and are designed specifically to facilitate the analysis of these results. For Windows and Linux.
- BioAdviser. Visualization tool for biomolecular structures and small molecules.
- iMolview. Application to browse and view in 3D protein and DNA structures from Protein Data Bank, and drug molecules from DrugBank For iPhone and iPad. Provided by Molsoft.
- PyMOL on the iPad.. High-performance 3D molecular visualizer, designed from the ground up for the iPad. it can search and download data from the PDB, PubChem, Dropbox, or an own secure custom PyMOL enterprise server. Provided by Schrödinger.
- RCSB PDB.. The RCSB Protein Data Bank (PDB) mobile app is the official mobile app of the RCSB PDB. It provides fast, on-the-go access to the RCSB PDB resources. The app enables the general public, researchers and scholars to search the Protein Data Bank and visualize protein structures using either a WiFi or cellular data connection.
- Ball&Stick. High-quality molecular visualization app for the iPad, iPhone and iPod Touch. Provided by MolySym.
- CueMol for iOS. Interactive macromolecular viewer for structural biologists. CueMol viewer allows the users to open and view the scene files made by the desktop version of CueMol, and the Protein Data Bank (PDB) format files, as well.
- 3D Molecules Edit&Drill. Application designed to enable students and professionals to build, construct, modify and examine molecules in 3D. Allows the users to open and view molecules in SDF format files, for example, from NCBI's PubChem. Developed by Virtualnye Prostranstva LLC.
- Chem3D for iPad. Chem3D for iPad enables scientists to view and manipulate 3D images of chemical and biochemical structures. Re-imagined for the iPad, the Chem3D app features a facile user interface to manipulate images using common touch, pinch and swipe gestures. Provided by PerkinElmer, Inc.
- CMol. Interactive 3D molecular viewer designed specifically for the iPad, iPhone and iPod touch. CMol allows the user to open and view PDB files with complete control over the representations and colours used for individual chains, residues and atoms.
- Molecules. Free application for iPhone and iPad, for viewing three-dimensional renderings of molecules and manipulating them using your fingers. You can rotate the molecules by moving your finger across the display, zoom in or out by using two-finger pinch gestures, or pan the molecule by moving two fingers across the screen at once. These structures can be viewed in both ball-and-stick and spacefilling visualization modes.
- iMolecular Builder. The IMoleBuilder is an application that can view, edit and build molecules in 3D. For iPhone.
- iPharosDreams. Molecular visualization app for iPad to perform in-silico drug discovery. Downloads protein structure files from Protein Data Bank, displays 3D molecules, touch, rotation, zoom in/out. Hierarchy structure of molecules is shown with a table that select components in a protein and related things. It can generate pharmacophores and analyze 3D protein-ligand interaction of biological macromolecules for in-silico drug discovery. Allows selecting a ligand from a protein and generate a binding site from the selected ligand. Can generate receptor based pharmacophores and get inspiration. Developed by EQUISnZAROO CO., LTD.
- Jmol Molecular Visualization. Free Jmol for Android tablets.
- NDKmol. Free molecular viewer for Android.
- Molecule Viewer 3D. Opens most common 3D molecule file formats saved on a SD card or found in a library of 243 included molecules. For Android.
- 3D Molecule View. 3D molecule viewer. For Android.
- Atomdroid. Free computational chemistry tool. It can be used as a molecular viewer/builder and contains local optimization and Monte Carlo simulation features. For Android.
- Atom 3D. Free application to visualize molecules and crystal structures in 3D using the touchscreen to rotate and zoom. Includes 19 sample structures. Supports XYZ files and some protein data bank (PDB) files. For Android.
- PDBs. Free application for molecular graphics visualization from PDB files. For Android.
- PDB View 3D. Application for molecular graphics visualization from PDB files. For Android.
Definitions and syntax of file formats
- Daylight SMILES. SMILES (Simplified Molecular Input Line Entry System) is a line notation (a typographical method using printable characters) for entering and representing molecules and reactions.
- InChI. (IUPAC International Chemical Identifier) is a string of characters capable of uniquely representing a chemical substance. It is derived from a structural representation of that substance in a way designed to be independent of the way that the structure was drawn (thus a single compound will always produce the same identifier). It provides a precise, robust, IUPAC approved tag for representing a chemical substance.
- Tripos Mol2. Complete description of the Mol2 file format (.mol2).
- PDB format. Complete description of the PDB file format (.pdb).
- SDF format. Complete description of the SDF file format (.sdf).
- SMARTS format. SMARTS Tutorial by Daylight.
- OpenSMILES. Community sponsored open-standards version of the SMILES language for chemistry. OpenSMILES is part of the Blue Obelisk community.
File format Converters
- OpenBabel. Free open source chemical expert system mainly used for converting chemical file formats. For Windows, Unix, and Mac OS.
- Indigo. Universal organic chemistry toolkit, containing tools for end users, as well as a documented API for developers. Free and open-source, but also available on a commercial basis. Distributed by GGA software.
- Indigo-depict. Command-line molecule and reaction rendering utility. Free and open source. Distibuted by GGA software.
- Indigo-cano. Command-line canonical SMILES generator. Free and open source. Distibuted by GGA software.
- Indigo-deco. Command-line program for R-Group deconvolution. Free and open source. Distibuted by GGA software.
- iBabel. iBabel is an alternative graphical interface to Open Babel for Macintosh OS X.
- PerlMol. Collection of perl modules providing objects and methods for representing molecules, atoms, and bonds in Perl; doing substructure matching; and reading and writing files in various formats.
Analysis of ligand-protein interactions
- PoseView. Automatically generates 2D structure-diagrams of protein-ligand complexes (png, svg and pdf) provided as 3D-input. Such input may come directly from crystal structures or be computed for example by a docking program. PoseView images are available for the majority of PDB-structures on the PDB web site. Developed by the University of Hamburg and distributed by BioSolveIT.
- PLiP. Web service and command line tool for fully automated characterization and analysis of non-covalent interactions between proteins and ligands in 3D structures. Developed by the Technische Universität of Desden, Germany.
- Ligplot+. Java interface of Ligplot, a program for automatic generation of 2D ligand-protein interaction diagrams. Developed and proposed free-for-non-profit by the European Bioinformatics Institute (EMBL-EBI).
- LeView. Java program that to generate 2D representations of ligands and their environments and binding interactions for PDB entries. It can be used automatically (in command line) or interactively (with a graphical interface). Provided free of charge by the Institut Pasteur de Lille, France.
- DS Visualizer. Free 3D visualizer of Discovery Studio. Allows sequence handling and, 2D or 3D charting. Creates 2D ligand-receptor interaction diagrams. Distributed by Accelrys. DS Visualizer ActiveX Control allows visualizing and interacting with molecules in Microsoft Office documents and Internet Explorer. For Windows and Linux.
- BINANA. (BINding ANAlyzer). Python-implemented algorithm for analyzing ligand binding. The program identifies key binding characteristics like hydrogen bonds, salt bridges, and pi interactions. As input, BINANA accepts receptor and ligand files in the PDBQT format. Allows visualization with VMD. Developed by the National Biomedical Computation Resource.
Web services
- iview. Interactive WebGL visualizer of protein-ligand complex. Developed by the Chinese university of Hong Kong.
- PoseView. Automatically generates 2D structure-diagrams of protein-ligand complexes provided as 3D-input. Such input may come directly from crystal structures or be computed for example by a docking program. Developed by the University of Hamburg and distributed by BioSolveIT.
- LCT. The Ligand Contact Tool calculates contacts between protein and ligand atoms, several parameters are available (distance cut-off, Van Der Waals radii usage, etc). Queries acepted are uploadable PDB format file or PDB accession code. Provided by the Structural Computational Biology Group of the Spanish national Cancer Research Centre.
- SimiCon. Identifies the equivalent protein-ligand atomic contacts between Reference and Target complexes. Results are shown as text, tables and 3D interactive graphics
Others
- ChemMobi. ChemMobi is a tool for Chemists, Biochemists and anyone else interested in chemical structures, chemical sourcing, chemical properties and safety information. For iPhone.
- ChemSpotlight. ChemSpotlight is a plugin for Mac OS X 10.5 and later, which reads common chemical formats and provides searching and preview in the Finder. ChemSpotlight reads common chemical file formats using the Open Babel chemistry library. Spotlight can then index and search chemical data: molecular weights, formulas, SMILES, InChI, fingerprints, etc. Developed by Geoffrey Hutchison. Free and open source.