Swiss Institute of Bioinformatics
Click2Drug
Directory of computer-aided Drug Design tools
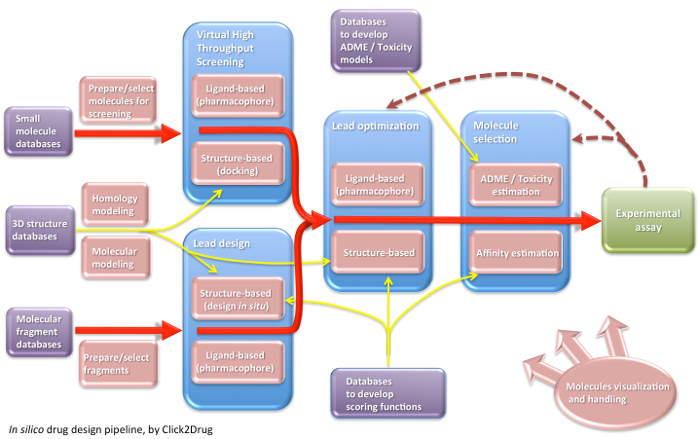
Click2Drug contains a comprehensive list of computer-aided drug design (CADD) software, databases and web services. These tools are classified according to their application field, trying to cover the whole drug design pipeline. If you think that an interesting tool is missing in this list, please contact usClick on the following picture to select tools related to a given activity:
Show all links Hide all links
Databases
Chemical databases
- Zinc Database. Curated collection of commercially available chemical compounds, with 3D coordinates, provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- Zinc15 Database. A new version of ZINC database including 100+ million purchasable compounds in ready-to-dock, 3D formats, provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- ChEMBL. Curated database of small molecules. Includes interactions and functional effects of small molecules binding to their macromolecular targets, and series of drug discovery databases.
- Chemspider. Collection of chemical compunds maintained by the Royal Society of Chemistry. Includes the conversion of chemical names to chemical structures, the generation of SMILES and InChI strings as well as the prediction of many physicochemical parameters.
- CoCoCo. Free suite of multiconformational molecular databases for High-Throughput Virtual Screening. It has single and multi conformer databases prepared for HTVS in different formats like Phase, Catalyst, Unity and SDF. Provided by the Department of Pharmaceutical Sciences of the University of Modena and Reggio Emilia.
- DrugBank. Bioinformatics and cheminformatics resource combining detailed drug (i.e. chemical, pharmacological and pharmaceutical) data with comprehensive drug target (i.e. sequence, structure, and pathway) information. Allows searching for similar compounds.
- PubChem. Database of chemical compounds maintained by the National Center for Biotechnology Information (NCBI), along with bioassays results. Allows similar compounds search (2D and 3D).
- TCM. Free small molecular database on traditional Chinese medicine, for virtual screening. It is currently the world's largest TCM database, and contains 170'000 compounds, with 3D mol2 and 2D cdx files, which passed ADMET filters.
- SCUBIDOO. a freely accessible database concept that currently holds 21 million virtual products originating from a small library of building blocks and a collection of robust organic reactions. This large data set was reduced to three representative and computationally tractable samples denoted as S, M, and L, containing 9994, 99 977, and 999 794 products, respectively. These small sets are useful as starting points for ligand identification and optimization projects. Proposed by the University of University of Marburg, Germany.
- Mcule database. Commercial database of commercially available small molecules. Allows filtering by chemical supplier data (stock availability, price, delivery time, chemical suppliers, catalogs, minimum purity, etc.) and export the whole Mcule database including supplier and procurement related properties. Reduced prices for academic. Provided by Mcule.
- WOMBAT. (World of Molecular Bioactivity). Database of 331,872 entries (268,246 unique SMILES), representing 1,966 unique targets, with bioactivity annotations. Compiled by Sunset Molecular Discovery LLC.
- Approved Drugs. The Approved Drugs app contains over a thousand chemical structures and names of small molecule drugs approved by the US Food & Drug Administration (FDA). Structures and names can be browsed in a list, searched by name, filtered by structural features, and ranked by similarity to a user-drawn structure. The detail view allows viewing of a 3D conformation as well as tautomers. Structures can be exported in a variety of ways, e.g. email, twitter, clipboard. For iPad and iPhone. Developed by Molecular Materials Informatics, Inc.
- ChemSpider Mobile. Allows searching the ChemSpider chemical database, provided by the Royal Society of Chemistry. Compounds can be searched by structure or by name, and browsed within the app. Results can be examined by jumping to the web page. Search structures are drawn using the powerful MMDS molecular diagram editor. For iPad. Provided by Molecular Materials Informatics, Inc.
- e-Drug3D. Database mirroring the current content of the U.S. pharmacopeia of small drugs. Contains 1822 molecular structures with a molecular weight < 2000 (last update: July 2016). Provides SD files (single conformer, tautomers or multiple conformers). Maintained by the Institut de Pharmacologie Moléculaire et Cellulaire, France.
- GLASS. GLASS (GPCR-Ligand Association) database is a manually curated repository for experimentally-validated GPCR-ligand interactions. Along with relevant GPCR and chemical information, GPCR-ligand association data are extracted and integrated into GLASS from literature and public databases. A list of currently-known GPCRs was compiled from UniProt and used to filter through the other chemical databases for ligand-association data (ChEMBL, BindingDB, IUPHAR, DrugBank, PDSP), GPCR diseases association (TTD), GPCR experimental structural data (PDB, BioLiP), and predicted models of GPCRs (GPCRRD). Subsequently, information from the extracted databases were unified to the same format and checked to ensure that all entries are only GPCR-related. Thus, the user would not find any entries on receptor tyrosine kinases or any other protein that is not a GPCR. All relevant ligand chemical data (PubChem) and GPCR data (UniProt) were extracted accordingly for each GPCR-ligand entry. Each molecule with a unique InChI key was considered a unique ligand entry in the database. Developed and maintained by the Zhang Lab at the University of Michigan, USA.
- ChemDB/ChemicalSearch. Find chemicals by various search criteria.
- Structural Database (CSD). Repository for small molecule crystal structures in CIF format. The CSD is compiled and maintained by the Cambridge Crystallographic Data Centre
- SPRESIweb. Integrated database containing over 8.7 million molecules, 4.1 million reactions, 658,000 references and 164,000 patents covering the years 1974 - 2009. Developed by InfoChem.
- MMsINC. Database of non-redundant, annotated and biomedically relevant chemical structures. Includes the analysis of chemical properties, such as ionization and tautomerization processes, and the in silico prediction of 24 important molecular properties in the biochemical profile of each structure. MMsINC supports various types of queries, including substructure queries and the novel 'molecular scissoring' query. MMsINC is interfaced with other primary data collectors, such as PubChem, Protein Data Bank (PDB), the Food and Drug Administration database of approved drugs and ZINC. provided by the CRS4 - Bioinformatics Laboratory, Parco Sardegna Ricerche, Italy.
- ZINClick. ZINClick is a database of triazoles generated using existing alkynes and azides, synthesizable in no more than three synthetic steps from commercially available products. This resulted in a combinatorial database of over 16 million of 1,4-disubstituted-1,2,3-triazoles (Molecular Weight < 1000), each of which is easily synthesizable, but at the same time new and patentable. Provided by the Università degli Studi del Piemonte Orientale "A. Avogadro".
- SPRESImobile. iPod, iPhone and iPad application providing direct access to ChemReact, a subset of the SPRESI structure and reaction database, which contains more than 400,000 unique reaction types and the related references. Developed by InfoChem.
- MORE. (MObile REagents). Mobile app, for iphone, ipad and android, which gives access to over 9 million molecules and 16 million chemical product variations offered by 56 different suppliers. Can search reagents by name, formula or by drawing a chemical structure. It is possible to limit the search to specific suppliers, bookmark the search results, and export small sdfiles. Allows converting a picture of a chemical structure taken from the iPhone camera into a structurally searchable molecule using OSRA (Optical Structure Recognition Application).
- KKB. (Kinase Knowledgebase). Database of kinase structure-activity and chemical synthesis data. Developed and maintained by Eidogen-Sertanty, Inc.
- iKinase Universal. iPad/iPhone application providing sample structure activity data from Eidogen-Sertanty's Kinase Knowledgebase (KKB). Exists in a Pro version (iKinasePro).
- DUD.E. (Database of Useful Decoys: Enhanced). DUD-E is designed to help test docking algorithms by providing challenging decoys. It contains a total 22,886 active compounds and their affinities against 102 targets, an average of 224 ligands per target. Also includes 50 decoys for each active, having similar physico-chemical properties but dissimilar 2-D topology. DUD-E is provided freely by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- DUD. (Directory of Useful Decoys). DUD is designed to help test docking algorithms by providing challenging decoys. It contains a total of 2,950 active compounds against a total of 40 targets. For each active, 36 "decoys" with similar physical properties (e.g. molecular weight, calculated LogP) but dissimilar topology. DUD is provided freely by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- GPCR-Bench. GPCR-Bench provides a high quality GPCR docking benchmarking set: 25 PDB structures covering all NR structures as of January 2015, and active and decoy compounds in the spirit of DUD. Provided by Heptares Therapeutics Ltd., UK.
- MUV. Maximum Unbiased Validation Datasets for Virtual Screening, with non-clumpy, spatially random topology. Provided by Carolo-Wilhelmina University.
- GLL. (GPCR Ligand Library). Database of 25145 ligands for 147 GPCRs. Associated with the GDD (GPCR Decoy Database). Provided by the Claudio N. Cavasotto Lab. of the Instituto de Biomedicina de Buenos Aires - Max Planck Society Partner (IBioBA-MPSP).
- GDD. (GPCR Decoy Database). For each ligand in GLL, 39 decoys were drawn from ZINC ensuring physical similarity of six properties (molecular weight, formal charge, hydrogen bond donors and acceptors, rotatable bonds and logP), but structural dissimilarity. Provided by the Claudio N. Cavasotto Lab. of the Instituto de Biomedicina de Buenos Aires - Max Planck Society Partner (IBioBA-MPSP).
- VDS. Virtual Decoy Sets for Molecular Docking Benchmarks. Similar to DUD but ignoring synthetic feasibility. Expected to be less biased with respect to physical similarity.
- LEADS-PEP. A benchmark dataset for assessing peptide docking performance. The set includes 53 protein-peptide complexes with peptide ranging from 3 to 12 residues. Several well-known small molecule docking program were tested. Provided by the Fraunhofer Institute for Molecular Biology and Applied Ecology, Germany.
- DNP. (Dictionary of Natural Products). Comprehensive and fully-edited database on natural products, arising from the Dictionary of Organic Compounds (DOC).The compilation of DNP is undertaken by a team of academics and freelancers who work closely with the in-house editorial staff at Chapman & Hall. Each contributor specialises in a particular natural product class (e.g. alkaloids) and reorganises and classifies the data in the light of new research so as to present it in the most consistent and logical manner possible.
- ChemIDPlus. Database of compounds and structures by US National Library of Medicine
- ChemBank. Public, web-based informatics environment created by the Broad Institute's Chemical Biology Program. Includes freely available data derived from small molecules and small-molecule screens, and resources for studying the data.
- eMolecules. Database of unique molecules from commercial suppliers
- GLIDA. GPCR-Ligand Database. Provides information on both GPCRs and their known ligands. Enterable either by GPCR search or ligand search. Maintained by the PharmacoInformatics Laboratory, Kyoto University.
- Comparative Toxicogenomics Database (CTD). Database of manually curated data describing cross-species chemical-gene/protein interactions and chemical and gene disease relationships to illuminate molecular mechanisms underlying variable susceptibility and environmentally influenced diseases.
- SuperDRUG2. Database of more than 4,600 active pharmaceutical ingredients. Annotations include drugs with regulatory details, chemical structures (2D and 3D), dosage, biological targets, physicochemical properties, external identifiers, side-effects and pharmacokinetic data. Different search mechanisms allow navigation through the chemical space of approved drugs. A 2D chemical structure search is provided in addition to a 3D superposition feature that superposes a drug with ligands already known to be found in the experimentally determined protein-ligand complexes. It has been added simulations of "physiologically-based" pharmacokinetics of drugs. The interaction check feature identifies potential drug-drug interactions and also provides alternative recommendations for elderly patients. Maintained by the University of Charité, Berlin, Germany.
- Ligand Expo. Formerly Ligand Depot. Provides chemical and structural information about small molecules within the structure entries of the Protein Data Bank.
- Glide Ligand Decoys Set. Collection created by selecting 1000 ligands from a one million compound library that were chosen to exhibit "drug-like" properties. Used in Glide enrichment studies. Provided by Schrödinger.
- Glide Fragment Library. Set of 441 unique small fragments (1-7 ionization/tautomer variants; 6-37 atoms; MW range 32-226) derived from molecules in the medicinal chemistry literature. The set includes a total of 667 fragments with accessible low energy ionization and tautomeric states and metal and state penalties for each compound from Epik. These can be used for fragment docking, core hopping, lead optimization, de novo design, etc. Provided by Schrödinger.
- Virtual library Repository. Libraries of 30,184 (redundant) and 4,544 small-molecule fragments, all less than 150 daltons in weight, derived from FDA-approved compounds using the python script fragmentizer. Distributed by the National Biomedical Computation Resource.
- NRDBSM. (Non Redundant Database of Small Molecules) is a database aimed specifically at virtual high throughput screening of small molecules. It has been developed giving special consideration to physicochemical properties and Lipinski's rule of five. Provided by the Supercomputing Facility for Bioinformatics & Computational Biology, IIT Delhi.
- Ligand Expo. Ligand Expo (formerly Ligand Depot) provides chemical and structural information about small molecules within the structure entries of the Protein Data Bank. Tools are provided to search the PDB dictionary for chemical components, to identify structure entries containing particular small molecules, and to download the 3D structures of the small molecule components in the PDB entry. A sketch tool is also provided for building new chemical definitions from reported PDB chemical components.
- ChEBI. (Chemical Entities of Biological Interest). Freely available dictionary of molecular entities focused on ‘small’ chemical compounds. provided by the European Bioinformatics Institute.
- KEGG DRUG. Comprehensive drug information resource for approved drugs in Japan, USA, and Europe unified based on the chemical structures and/or the chemical components, and associated with target, metabolizing enzyme, and other molecular interaction network information. Provided by the Kyoto Encyclopedia of Genes and Genomes.
Databases handling
- Bingo. Relational database management system (RDBMS) data cartridge that provides fast, scalable, and efficient storage and searching solution for chemical information. Bingo integrates the chemistry into Oracle, Microsoft SQL Server and PostgreSQL databases. Its extensible indexing is designed to enable scientists to store, index, and search chemical moieties alongside numbers and text within one underlying relational database server. Free software. Distributed by GGA software.
- JChem for Excel. Integrates structure handling and visualizing capabilities within a Microsoft Excel environment. Structures are fully supported within spreadsheets and be can viewed, edited, searched, resized, ordered, managed. Provided by ChemAxon.
- ChemDiff. Indigo-based utility for finding duplications and visual comparison of two files containing multiple structures. SDF, SMILES, CML, MOLFILE input formats are supported. Files can contains large amount of molecules and ChemDiff was test on files with up to 1 million ones. Free and open-source. Distributed by GGA software.
- IXTAB. Xtab is a transversal compounds library management tool to create, import, explore and analyse databases. Provided by Mind The Byte.
Screening
Web services
- e-LEA3D. Searches the FDA approved drugs either by keyword or by substructure. Also builds combinatorial library of molecules.
- Combinatorial library design. Web server providing a click chemistry engine to connect one or more reactants on a central core (scaffold).
- eDesign. Web server providing a de novo drug design engine to create new molecules either from scratch (lead-hopping) or based on a user-defined scaffold on which R-groups have to be optimized. Alternatively, the same tool can be used to screen a library of molecules. The sructure-based function is based on the program PLANTS. Maintained by the Institut de Pharmacologie Moléculaire et Cellulaire, France.
- GFscore. Web server to discriminate true negatives from false negatives in a dataset of diverse chemical compounds using a consensus scoring in a Non-Linear Neural Network manner. The global scoring function is a combination of the five scoring functions found in the Cscore package from Tripos Inc.
- wwLig-CSRre. Online Tool to enrich a bank a small compound with compounds similar to a query.
Ligand design
Web services
- eDesign. Web server providing a de novo drug design engine to create new molecules either from scratch (lead-hopping) or based on a user-defined scaffold on which R-groups have to be optimized. Alternatively, the same tool can be used to screen a library of molecules. The structure-based function is based on the program PLANTS. Maintained by the Institut de Pharmacologie Moléculaire et Cellulaire, France.