Swiss Institute of Bioinformatics
Click2Drug
Directory of computer-aided Drug Design tools
Click2Drug contains a comprehensive list of computer-aided drug design (CADD) software, databases and web services. These tools are classified according to their application field, trying to cover the whole drug design pipeline. If you think that an interesting tool is missing in this list, please contact usClick on the following picture to select tools related to a given activity:
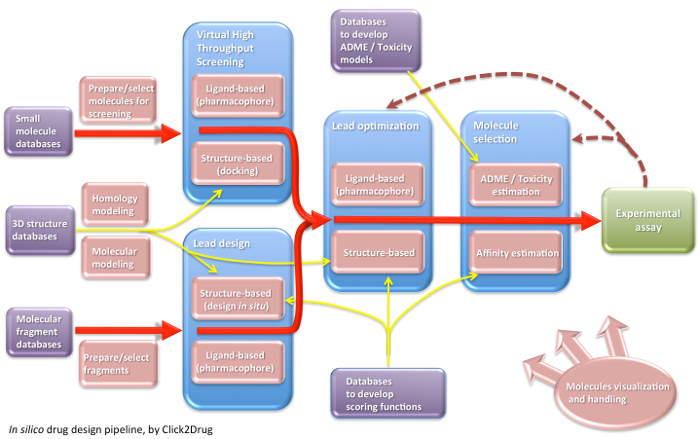
Show all links Hide all links
Target prediction
Software
- PatchSearch. PatchSearch implements local searching for similar binding sites on protein surfaces with a controlled amount of flexibility. It is based on product graphs to represent all possible matchings between two structures. Developed and provided as an R package by Univeristé Paris-Diderot, France.
- IXCHEL. Ixchel is a protein-based biological activity prediction application. The input molecule (in SDF or SMILE) is docked in a database of over 9 000 protein cavities. Distributed by Mind The Byte.
- CABRAKAN. Cabrakan is a 2D ligand-based virtual profiling application. It compares molecules through their 2D-fingerprints and predicts their biological activity. Distributed by Mind The Byte.
- HURAKAN. Hurakan is a 3D ligand-based virtual profiling application. It compares molecules according to their interaction with their environment, without superimposition, to obtain compounds with different structures but predicted with similar bioactivity. Distributed by Mind The Byte.
- MolScore-Antivirals. Expert system to identify and prioritise antiviral drug candidates. Developed by PharmaInformatic, Germany.
- MolScore-Antibiotics. Expert system to identify and prioritise antibacterial drug candidates. Developed by PharmaInformatic, Germany.
Web services
- SwissTargetPrediction. Online tool to predict the targets of bioactive small molecules in human and other vertebrates. This is useful to understand the molecular mechanisms underlying a given phenotype or bioactivity, to rationalize possible side-effects or to predict off-targets of known molecules. Provided by the Molecular Modeling group of the Swiss Institute of BioInformatics.
- SEA. SEA (Similarity ensemble approach) relates proteins based on the set-wise chemical similarity among their ligands. It can be used to rapidly search large compound databases, build cross-target similarity maps and predict possible targets of a small molecule. Provided by the Shoichet Laboratory in the Department of Pharmaceutical Chemistry at the University of California, San Francisco (UCSF).
- CSNAP. CSNAP (Chemical Similarity Network Analysis Pull-down) is a computational approach for compound target identification based on network similarity graphs. Query and reference compounds are populated on the network connectivity map and a graph-based neighbor counting method is applied to rank the consensus targets among the neighborhood of each query ligand. Developed in the Torres lab at the University of California, Los Angeles (UCLA).
- PPB. PPB (Polypharmacology Browser) searches through 4613 groups of at least 10 bioactive molecules with documented activity against a biological target, as listed in ChEMBL, to identify analogs of any query molecule using six different fingerprints and four fingerprint combination (HyperSpace), and displays results groups by targets as lists of bioactive compounds. Provided by the Dept. of Chemistry and Biochemistry of the University of Bern, Switzerland.
- ChemProt. The ChemProt 2.0 server is a ressource of annotated and predicted chemical-protein interactions. The server is a compilation of over 1 100 000 unique chemicals with biological activity for more than 15000 proteins. ChemProt can assist in the in silico evaluation of small molecules (drugs, environmental chemicals and natural products) with the integration of molecular, cellular and disease-associated proteins complexes. Provided by the Technical University of Denmark, and the University Paris Diderot.
- SuperPred. Webservice for drug classification and target prediction. The web-server translates a user-defined molecule into a structural fingerprint that is compared to about 6300 drugs, which are enriched by 7300 links to molecular targets of the drugs, derived through text mining followed by manual curation. Provided by the Institute of Molecular Biology and Bioinformatics, Charité - University Medicine Berlin.
- PASSonline. (Prediction of Activity Spectra for Substances). Web service for evaluating the general biological potential of an organic drug-like molecule, based on the comparison of the user's compound to a database of 260,000 of drug-like biologically active compounds using the Multilevel Neighborhoods of Atoms (MNA) structure descriptors. Provided by the Orekhovich Institute of Biomedical Chemistry
- Target Hunter of Small Molecule. Web portal for predicting the therapeutic potential of small organic molecules based on chemogenomic database. Created and maintained by Prof. Xiang-Qun (Sean) Xie’s laboratory
- HitPick. Web server that facilitates the analysis of chemical screenings by identifing hits and predicting their molecular targets. For target prediction, HitPick applies an approach that combines two 2D molecular similarity based methods: a simple 1-Nearest-Neighbour similarity searching and a machine learning method based on Laplacian-modified naive Bayesian models. provided by the Helmholtz Center Munich, germany.
- Molinspiration bioactivity score. Score a compound for its ability to be GPCR ligand, ion channel modulator, kinase inhibitor, nuclear receptor ligand, protease inhibitor, enzyme inhibitor. Based on Bayesian statistics to compare structures of representative ligands active on the particular target with structures of inactive molecules and to identify substructure features (which in turn determine physicochemical properties) typical for active molecules. Provided by Molinspiration.
- ElectroShape Polypharmacology server. Web service to estimate polypharmacology profiles and side effects of compounds based on the molecular similarity concept. Developed and maintained by Alvaro Cortes Cabrera.